gget


![]()
gget is a free and open-source command-line tool and Python package that enables efficient querying of genomic databases. gget consists of a collection of separate but interoperable modules, each designed to facilitate one type of database querying in a single line of code.
Please cite the following paper:
Luebbert, L. & Pachter, L. (2022). Efficient querying of genomic databases for single-cell RNA-seq with gget. bioRxiv 2022.05.17.492392; doi: https://doi.org/10.1101/2022.05.17.492392
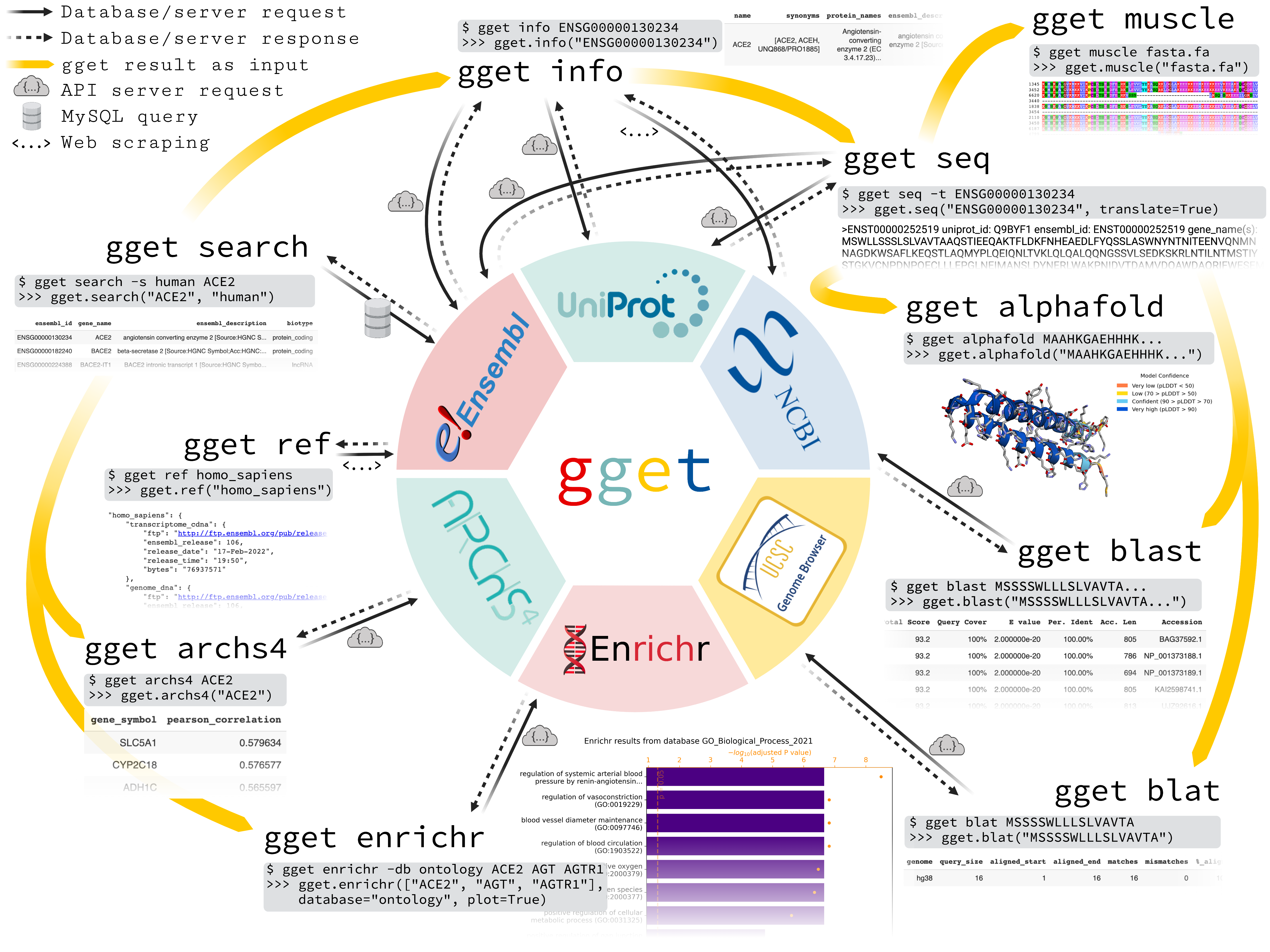
gget currently consists of the following nine modules:
gget ref
Fetch File Transfer Protocols (FTPs) and metadata for reference genomes and annotations from Ensembl by species.gget search
Fetch genes and transcripts from Ensembl using free-form search terms.gget info
Fetch extensive gene and transcript metadata from Ensembl, UniProt, and NCBI using Ensembl IDs.gget seq
Fetch nucleotide or amino acid sequences of genes or transcripts from Ensembl or UniProt, respectively.gget blast
BLAST a nucleotide or amino acid sequence to any BLAST database.gget blat
Find the genomic location of a nucleotide or amino acid sequence using BLAT.gget muscle
Align multiple nucleotide or amino acid sequences to each other using Muscle5.gget enrichr
Perform an enrichment analysis on a list of genes using Enrichr.gget archs4
Find the most correlated genes to a gene of interest or find the gene's tissue expression atlas using ARCHS4.
Installation
pip install gget
For use in Jupyter Lab / Google Colab:
import gget
Quick start guide
# Fetch all Homo sapiens reference and annotation FTPs from the latest Ensembl release
$ gget ref -s homo_sapiens
# Search human genes with "ace2" AND "angiotensin" in their name/description
$ gget search -sw ace2,angiotensin -s homo_sapiens -ao and
# Look up gene ENSG00000130234 (ACE2) with expanded info (returns all transcript isoforms for genes)
$ gget info -id ENSG00000130234 -e
# Fetch the amino acid sequence of the canonical transcript of gene ENSG00000130234
$ gget seq -id ENSG00000130234 --seqtype transcript
# Quickly find the genomic location of (the start of) that amino acid sequence
$ gget blat -seq MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS
# Blast (the start of) that amino acid sequence
$ gget blast -seq MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS
# Align nucleotide or amino acid sequences stored in a FASTA file
$ gget muscle -fa path/to/file.fa
# Use Enrichr to find the ontology of a list of genes
$ gget enrichr -g ACE2 AGT AGTR1 ACE AGTRAP AGTR2 ACE3P -db ontology
# Get the human tissue expression atlas of gene ACE2
$ gget archs4 -g ACE2 -w tissue
Jupyter Lab / Google Colab:
gget.ref("homo_sapiens")
gget.search(["ace2", "angiotensin"], "homo_sapiens", andor="and")
gget.info("ENSG00000130234", expand=True)
gget.seq("ENSG00000130234", seqtype="transcript")
gget.blat("MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS")
gget.blast("MSSSSWLLLSLVAVTAAQSTIEEQAKTFLDKFNHEAEDLFYQSSLAS")
gget.muscle("path/to/file.fa")
gget.enrichr(["ACE2", "AGT", "AGTR1", "ACE", "AGTRAP", "AGTR2", "ACE3P"], database="ontology", plot=True)
gget.archs4("ACE2", which="tissue")
Manual
Jupyter Lab / Google Colab arguments are equivalent to long-option arguments (--arg).
The manual for any gget tool can be called from terminal using the -h --help flag.
gget ref
Fetch FTPs and their respective metadata (or use flag ftp to only return the links) for reference genomes and annotations from Ensembl by species.
Return format: dictionary/json.
Required arguments
-s --species
Species for which the FTPs will be fetched in the format genus_species, e.g. homo_sapiens.
Note: Not required when calling flag [--list_species].
Supported shortcuts: 'human', 'mouse'
Optional arguments
-w --which
Defines which results to return. Default: 'all' -> Returns all available results.
Possible entries are one or a combination of the following:
'gtf' - Returns the annotation (GTF).
'cdna' - Returns the trancriptome (cDNA).
'dna' - Returns the genome (DNA).
'cds' - Returns the coding sequences corresponding to Ensembl genes. (Does not contain UTR or intronic sequence.)
'cdrna' - Returns transcript sequences corresponding to non-coding RNA genes (ncRNA).
'pep' - Returns the protein translations of Ensembl genes.
-r --release
Defines the Ensembl release number from which the files are fetched, e.g. 104. Default: latest Ensembl release.
-o --out
Path to the json file the results will be saved in, e.g. path/to/directory/results.json. Default: Standard out.
Jupyter Lab / Google Colab: save=True will save the output in the current working directory.
Flags
-l --list_species
Lists all available species. (Jupyter Lab / Google Colab: combine with species=None.)
-ftp --ftp
Returns only the requested FTP links.
-d --download
Downloads the requested FTPs to the current directory (requires curl to be installed).
Examples
Use gget ref in combination with kallisto | bustools to build a reference index:
kb ref -i INDEX -g T2G -f1 FASTA $(gget ref --ftp -w dna,gtf -s homo_sapiens)
→ kb ref builds a reference index using the latest DNA and GTF files of species Homo sapiens passed to it by gget ref.
Get all available genomes:
gget ref --list -r 103
# Jupyter Lab / Google Colab:
gget.ref(species=None, list_species=True, release=103)
→ Returns a list with all available genomes (checks if GTF and FASTAs are available) from Ensembl release 103.
(If no release is specified, gget ref will always return information from the latest Ensembl release.)
Get the genome reference for a specific species:
gget ref -s homo_sapiens -w gtf dna
# Jupyter Lab / Google Colab:
gget.ref("homo_sapiens", which=["gtf", "dna"])
→ Returns a json with the latest human GTF and FASTA FTPs, and their respective metadata, in the format:
{
"homo_sapiens": {
"annotation_gtf": {
"ftp": "http://ftp.ensembl.org/pub/release-106/gtf/homo_sapiens/Homo_sapiens.GRCh38.106.gtf.gz",
"ensembl_release": 106,
"release_date": "28-Feb-2022",
"release_time": "23:27",
"bytes": "51379459"
},
"genome_dna": {
"ftp": "http://ftp.ensembl.org/pub/release-106/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz",
"ensembl_release": 106,
"release_date": "21-Feb-2022",
"release_time": "09:35",
"bytes": "881211416"
}
}
}
More examples
gget search
Fetch genes and transcripts from Ensembl using free-form search terms.
Return format: data frame.
Required arguments
-sw --searchwords
One or more free form search words, e.g. gaba, nmda. (Note: Search is not case-sensitive.)
-s --species
Species or database to be searched.
A species can be passed in the format 'genus_species', e.g. 'homo_sapiens'.
To pass a specific database, pass the name of the CORE database, e.g. 'mus_musculus_dba2j_core_105_1'.
All availabale databases can be found here.
Supported shortcuts: 'human', 'mouse'.
Optional arguments
-st --seqtype
'gene' (default) or 'transcript'
Returns genes or transcripts, respectively.
-ao --andor
'or' (default) or 'and'
'or': Returns all genes that INCLUDE AT LEAST ONE of the searchwords in their name/description.
'and': Returns only genes that INCLUDE ALL of the searchwords in their name/description.
-l --limit
Limits the number of search results, e.g. 10. Default: None.
-o --out
Path to the csv the results will be saved in, e.g. path/to/directory/results.csv. Default: Standard out.
Jupyter Lab / Google Colab: save=True will save the output in the current working directory.
Flags
wrap_text
Jupyter Lab / Google Colab only. wrap_text=True displays data frame with wrapped text for easy reading (default: False).
Example
gget search -sw gaba gamma-aminobutyric -s homo_sapiens
# Jupyter Lab / Google Colab:
gget.search(["gaba", "gamma-aminobutyric"], "homo_sapiens")
→ Returns all genes that contain at least one of the search words in their name or Ensembl/external reference description:
| ensembl_id | gene_name | ensembl_description | ext_ref_description | biotype | url |
|---|---|---|---|---|---|
| ENSG00000034713 | GABARAPL2 | GABA type A receptor associated protein like 2 [Source:HGNC Symbol;Acc:HGNC:13291] | GABA type A receptor associated protein like 2 | protein_coding | https://uswest.ensembl.org/homo_sapiens/Gene/Summary?g=ENSG00000034713 |
| . . . | . . . | . . . | . . . | . . . | . . . |
More examples
gget info
Fetch extensive gene and transcript metadata from Ensembl, UniProt, and NCBI using Ensembl IDs.
Return format: data frame.
Required arguments
-id --ens_ids
One or more Ensembl IDs.
Optional arguments
-o --out
Path to the csv the results will be saved in, e.g. path/to/directory/results.csv. Default: Standard out.
Jupyter Lab / Google Colab: save=True will save the output in the current working directory.
Flags
-e --expand
Expands returned information (only for gene and transcript IDs).
For genes, adds information on all known transcripts.
For transcripts, adds information on all known translations and exons.
wrap_text
Jupyter Lab / Google Colab only. wrap_text=True displays data frame with wrapped text for easy reading (default: False).
Example
gget info -id ENSG00000034713 ENSG00000104853 ENSG00000170296 -e
# Jupyter Lab / Google Colab:
gget.info(["ENSG00000034713", "ENSG00000104853", "ENSG00000170296"], expand=True)
→ Returns extensive information about each requested Ensembl ID in data frame format:
| uniprot_id | ncbi_gene_id | primary_gene_name | synonyms | protein_names | ensembl_description | uniprot_description | ncbi_description | biotype | canonical_transcript | ... | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ENSG00000034713 | P60520 | 11345 | GABARAPL2 | [ATG8, ATG8C, FLC3A, GABARAPL2, GATE-16, GATE16, GEF-2, GEF2] | Gamma-aminobutyric acid receptor-associated protein like 2 (GABA(A) receptor-associated protein-like 2)... | GABA type A receptor associated protein like 2 [Source:HGNC Symbol;Acc:HGNC:13291] | FUNCTION: Ubiquitin-like modifier involved in intra- Golgi traffic (By similarity). Modulates intra-Golgi transport through coupling between NSF activity and ... | Enables ubiquitin protein ligase binding activity. Involved in negative regulation of proteasomal protein catabolic process and protein... | protein_coding | ENST00000037243.7 | ... |
| . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | ... |
More examples
gget seq
Fetch nucleotide or amino acid sequence of a gene (and all its isoforms) or a transcript by Ensembl ID.
Return format: FASTA.
Required arguments
-id --ens_ids
One or more Ensembl IDs.
Optional arguments
-st --seqtype
'gene' (default) or 'transcript'.
Defines whether nucleotide or amino acid sequences are returned.
Nucleotide sequences are fetched from Ensembl.
Amino acid sequences are fetched from UniProt.
-o --out
Path to the file the results will be saved in, e.g. path/to/directory/results.fa. Default: Standard out.
Jupyter Lab / Google Colab: save=True will save the output in the current working directory.
Flags
-i --isoforms
Returns the sequences of all known transcripts.
(Only for gene IDs in combination with seqtype=transcript.)
Examples
gget seq -id ENSG00000034713 ENSG00000104853 ENSG00000170296
# Jupyter Lab / Google Colab:
gget.seq(["ENSG00000034713", "ENSG00000104853", "ENSG00000170296"])
→ Returns the nucleotide sequences of ENSG00000034713, ENSG00000104853, and ENSG00000170296 in FASTA format.
gget seq -id ENSG00000034713 -st transcript -iso
# Jupyter Lab / Google Colab:
gget.seq("ENSG00000034713", seqtype="transcript", isoforms=True)
→ Returns the amino acid sequences of all known transcripts of ENSG00000034713 in FASTA format.
More examples
gget blast
BLAST a nucleotide or amino acid sequence to any BLAST database.
Return format: data frame.
Required arguments
-seq --sequence
Nucleotide or amino acid sequence, or path to FASTA or .txt file.
Optional arguments
-p --program
'blastn', 'blastp', 'blastx', 'tblastn', or 'tblastx'.
Default: 'blastn' for nucleotide sequences; 'blastp' for amino acid sequences.
-db --database
'nt', 'nr', 'refseq_rna', 'refseq_protein', 'swissprot', 'pdbaa', or 'pdbnt'.
Default: 'nt' for nucleotide sequences; 'nr' for amino acid sequences.
More info on BLAST databases
-l --limit
Limits number of hits to return. Default: 50.
-e --expect
Defines the expect value cutoff. Default: 10.0.
-o --out
Path to the csv the results will be saved in, e.g. path/to/directory/results.csv. Default: Standard out.
Jupyter Lab / Google Colab: save=True will save the output in the current working directory.
Flags
-lcf --low_comp_filt
Turns on low complexity filter.
-mbo --megablast_off
Turns off MegaBLAST algorithm. Default: MegaBLAST on (blastn only).
-q --quiet
Prevents progress information from being displayed.
wrap_text
Jupyter Lab / Google Colab only. wrap_text=True displays data frame with wrapped text for easy reading (default: False).
Example
gget blast -seq MKWMFKEDHSLEHRCVESAKIRAKYPDRVPVIVEKVSGSQIVDIDKRKYLVPSDITVAQFMWIIRKRIQLPSEKAIFLFVDKTVPQSR
# Jupyter Lab / Google Colab:
gget.blast("MKWMFKEDHSLEHRCVESAKIRAKYPDRVPVIVEKVSGSQIVDIDKRKYLVPSDITVAQFMWIIRKRIQLPSEKAIFLFVDKTVPQSR")
→ Returns the BLAST result of the sequence of interest in data frame format. gget blast automatically detects this sequence as an amino acid sequence and therefore sets the BLAST program to blastp with database nr.
| Description | Scientific Name | Common Name | Taxid | Max Score | Total Score | Query Cover | ... |
|---|---|---|---|---|---|---|---|
| PREDICTED: gamma-aminobutyric acid receptor-as... | Colobus angolensis palliatus | NaN | 336983 | 180 | 180 | 100% | ... |
| . . . | . . . | . . . | . . . | . . . | . . . | . . . | ... |
BLAST from .fa or .txt file:
gget blast -seq fasta.fa
# Jupyter Lab / Google Colab:
gget.blast("fasta.fa")
→ Returns the BLAST results of the first sequence contained in the fasta.fa file.
More examples
gget blat
Find the genomic location of a nucleotide or amino acid sequence using BLAT.
Return format: data frame.
Required arguments
-seq --sequence
Nucleotide or amino acid sequence, or path to FASTA or .txt file.
Optional arguments
-st --seqtype
'DNA', 'protein', 'translated%20RNA', or 'translated%20DNA'.
Default: 'DNA' for nucleotide sequences; 'protein' for amino acid sequences.
-a --assembly
'human' (hg38) (default), 'mouse' (mm39), 'zebrafinch' (taeGut2),
or any of the species assemblies available here (use short assembly name).
-o --out
Path to the csv the results will be saved in, e.g. path/to/directory/results.csv. Default: Standard out.
Jupyter Lab / Google Colab: save=True will save the output in the current working directory.
Example
gget blat -seq MKWMFKEDHSLEHRCVESAKIRAKYPDRVPVIVEKVSGSQIVDIDKRKYLVPSDITVAQFMWIIRKRIQLPSEKAIFLFVDKTVPQSR -a taeGut2
# Jupyter Lab / Google Colab:
gget.blat("MKWMFKEDHSLEHRCVESAKIRAKYPDRVPVIVEKVSGSQIVDIDKRKYLVPSDITVAQFMWIIRKRIQLPSEKAIFLFVDKTVPQSR", assembly="taeGut2")
→ Returns BLAT results for assembly taeGut2 (zebra finch) in data frame format. In the above example, gget blat automatically detects this sequence as an amino acid sequence and therefore sets the BLAT seqtype to protein.
| genome | query_size | aligned_start | aligned_end | matches | mismatches | %_aligned | ... |
|---|---|---|---|---|---|---|---|
| taeGut2 | 88 | 12 | 88 | 77 | 0 | 87.5 | ... |
More examples
gget muscle
Align multiple nucleotide or amino acid sequences to each other using Muscle5.
Return format: ClustalW formatted standard out or aligned FASTA.
Required arguments
-fa --fasta
Path to FASTA or .txt file containing the nucleotide or amino acid sequences to be aligned.
Optional arguments
-o --out
Path to the aligned FASTA file the results will be saved in, e.g. path/to/directory/results.afa. Default: Standard out.
Jupyter Lab / Google Colab: save=True will save the output in the current working directory.
Flags
-s5 --super5
Aligns input using the Super5 algorithm instead of the Parallel Perturbed Probcons (PPP) algorithm to decrease time and memory.
Use for large inputs (a few hundred sequences).
wrap_text
Jupyter Lab / Google Colab only. wrap_text=True displays data frame with wrapped text for easy reading (default: False).
Example
gget muscle -fa fasta.fa
# Jupyter Lab / Google Colab:
gget.muscle("fasta.fa")
→ Returns an overview of the aligned sequences with ClustalW coloring. (To return an aligned FASTA (.afa) file, use --out argument (or save=True in Jupyter Lab/Google Colab).) In the above example, the 'fasta.fa' includes several sequences to be aligned (e.g. isoforms returned from gget seq).

More examples
gget enrichr
Perform an enrichment analysis on a list of genes using Enrichr.
Return format: data frame.
Required arguments
-g --genes
Short names (gene symbols) of genes to perform enrichment analysis on, e.g. 'PHF14 RBM3 MSL1 PHF21A'.
-db --database
Database to use as reference for the enrichment analysis.
Supports any database listed here under 'Gene-set Library' or one of the following shortcuts:
'pathway' (KEGG_2021_Human)
'transcription' (ChEA_2016)
'ontology' (GO_Biological_Process_2021)
'diseases_drugs' (GWAS_Catalog_2019)
'celltypes' (PanglaoDB_Augmented_2021)
'kinase_interactions' (KEA_2015)
Optional arguments
-o --out
Path to the csv the results will be saved in, e.g. path/to/directory/results.csv. Default: Standard out.
Jupyter Lab / Google Colab: save=True will save the output in the current working directory.
Flags
plot
Jupyter Lab / Google Colab only. plot=True provides a graphical overview of the first 15 results (default: False).
Example
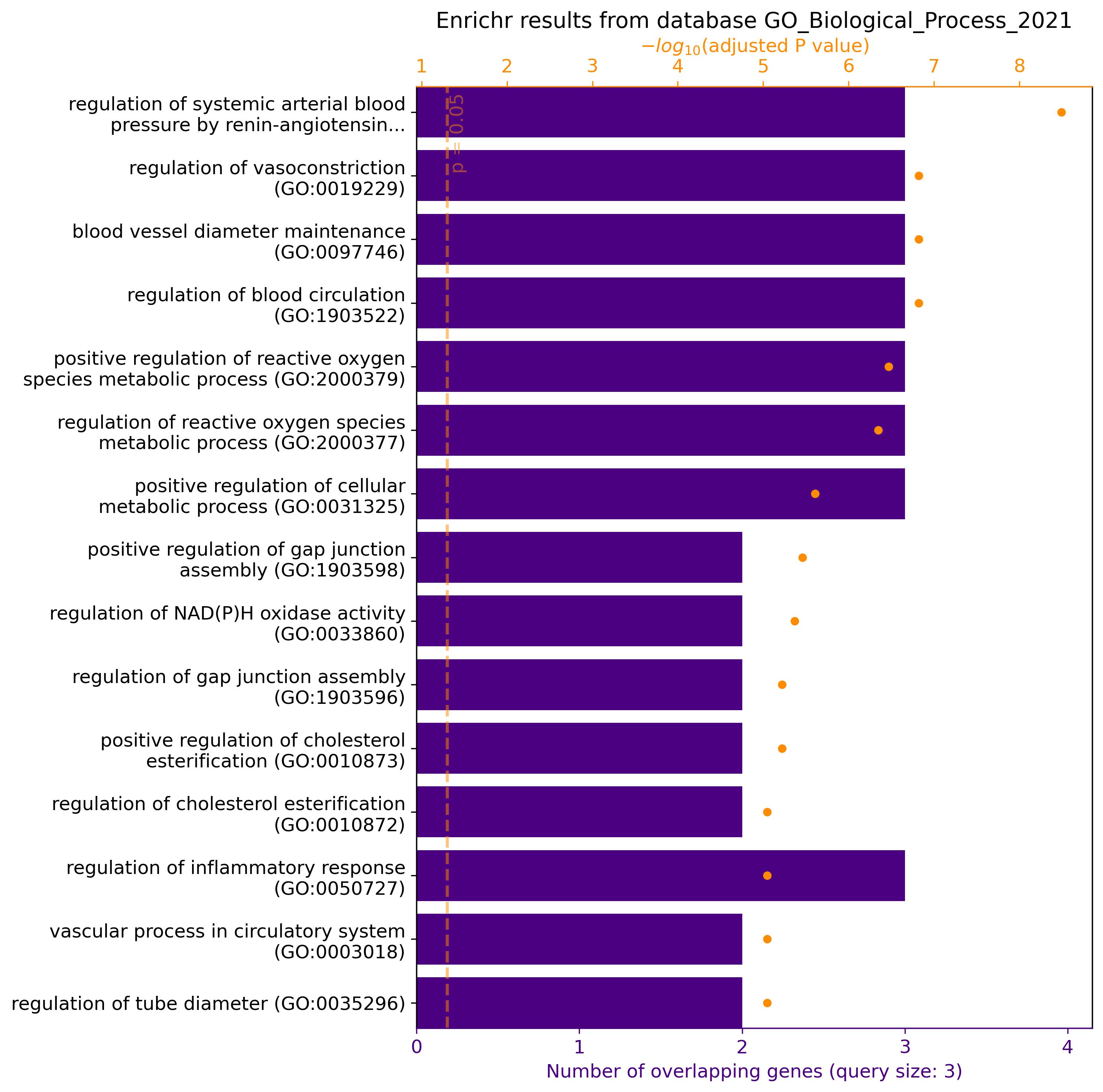
gget enrichr -g ACE2 AGT AGTR1 -db ontology
# Jupyter Lab / Google Colab:
gget.enrichr(["ACE2", "AGT", "AGTR1"], database="ontology", plot=True)
→ Returns pathways/functions involving genes ACE2, AGT, and AGTR1 from the GO Biological Process 2021 database in data frame format. In Jupyter Lab / Google Colab, plot=True returns a graphical overview of the results:
More examples
gget archs4
Find the most correlated genes to a gene of interest or find the gene's tissue expression atlas using ARCHS4.
Return format: data frame.
Required arguments
-g --gene
Short name (gene symbol) of gene of interest, e.g. 'STAT4'.
Optional arguments
-w --which
'correlation' (default) or 'tissue'.
'correlation' returns a gene correlation table that contains the 100 most correlated genes to the gene of interest. The Pearson correlation is calculated over all samples and tissues in ARCHS4.
'tissue' returns a tissue expression atlas calculated from human or mouse samples (as defined by 'species') in ARCHS4.
-s --species
'human' (default) or 'mouse'.
Defines whether to use human or mouse samples from ARCHS4.
(Only for tissue expression atlas.)
-o --out
Path to the csv the results will be saved in, e.g. path/to/directory/results.csv. Default: Standard out.
Jupyter Lab / Google Colab: save=True will save the output in the current working directory.
Examples
gget archs4 -g ACE2
# Jupyter Lab / Google Colab:
gget.archs4("ACE2")
→ Returns the 100 most correlated genes to ACE2 in a data frame:
| gene_symbol | pearson_correlation |
|---|---|
| SLC5A1 | 0.579634 |
| CYP2C18 | 0.576577 |
| . . . | . . . |
gget archs4 -g ACE2 -w tissue
# Jupyter Lab / Google Colab:
gget.archs4("ACE2", which="tissue")
→ Returns the tissue expression of ACE2 in a data frame (by default, human data is used):
| id | min | q1 | median | q3 | max |
|---|---|---|---|---|---|
| System.Urogenital/Reproductive System.Kidney.RENAL CORTEX | 0.113644 | 8.274060 | 9.695840 | 10.51670 | 11.21970 |
| System.Digestive System.Intestine.INTESTINAL EPITHELIAL CELL | 0.113644 | 5.905560 | 9.570450 | 13.26470 | 13.83590 |
| . . . | . . . | . . . | . . . | . . . | . . . |



















 13 Nov 28, 2022
13 Nov 28, 2022
 30 Sep 8, 2022
30 Sep 8, 2022
 75 Dec 4, 2022
75 Dec 4, 2022
 976 Jan 3, 2023
976 Jan 3, 2023
 17 Dec 11, 2022
17 Dec 11, 2022
 11 Aug 10, 2022
11 Aug 10, 2022
 0 Aug 10, 2021
0 Aug 10, 2021
 13 Nov 8, 2022
13 Nov 8, 2022
 14.5k Jan 3, 2023
14.5k Jan 3, 2023
 37 Dec 15, 2022
37 Dec 15, 2022
 18.6k Dec 30, 2022
18.6k Dec 30, 2022
 4 Sep 4, 2021
4 Sep 4, 2021
 22 Dec 5, 2022
22 Dec 5, 2022
 15 Jan 7, 2022
15 Jan 7, 2022
 5.8k Jan 3, 2023
5.8k Jan 3, 2023
 5 Dec 27, 2022
5 Dec 27, 2022
 1.3k Dec 28, 2022
1.3k Dec 28, 2022
 446 Dec 21, 2022
446 Dec 21, 2022
 112 Nov 5, 2022
112 Nov 5, 2022